The work in this
ACIEE EarlyView article stemmed from synthetic studies toward some polyketide-derived natural products, such as seragakinone A (
1) and the antibiotic BE-43472A (
2). A general method for installing quaternary stereogenic centers at the angular position of these natural products was required.

A general approach to solve this problem involved a pinacol type alkyl shift as shown in Scheme 2.
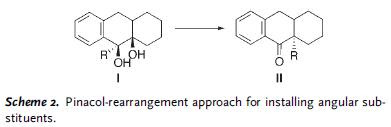
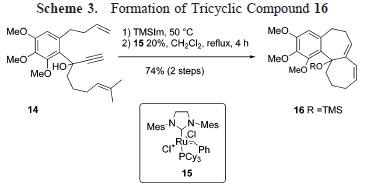
However, this approach had some issues that needed addressing. First, I needed to be generated stereoselectively. Second, since both carbinol carbons are tertiary, the OH at the angular position had to be the only leaving group to facilitate migration of R group. A successful two-step process was reported in this article, employing a substrate of type 3, incorporating oxazole ring in the tricyclic core. (R)-3 can be readily prepared in enantio-pure form. General strategy is summarized in Scheme 3.
It was found that starting with (R)-3, addition of an alkyl group proceeded stereoselectively to give cis-diol without the loss of ee, as seen in Scheme 4 in the case of vinyl addition. Pleasingly, treatment of 4a afforded enantio-enriched (S)-5a in high yield and high ee (no loss of ee from chiral 3).

When reaction started with starting material trans-diol 6, it was surprising to find that enantiomeric (R)-5a was obtained in excellent yield and high ee.

This result illustrated the amazing carbocation-stabilizing ability of the oxazole ring. This concept was and illustrated again in separate racemization experiments as shown in equations 2 and 3.

As shown in Eq 2 and 3, when chiral (R)-3 was subjected to protic acid conditions, 3 was recovered with only 60% ee in 94% yield. And when (R)-3 was subjected to Lewis acid-promoted allylsilane addition, 5e was afforded in racemic form. These two experiments showed that racemization occurred in the generation of carbocation at the carbinol carbon, stabilized by oxazole. This is particularly impressive provided that this carbocation was situated alpha to a carbonyl.
The overall mechanism of the process was proposed and summarized in Scheme 5. Basically, exposure of cis-diol to Lewis acid could lead to intermediate A, which led to enantio-enriched product C, or racemization intermediate B, which ultimately led to ent-C.
Having established the method, several substrates were investigated for scope of the reaction. All pinacol rearrangement substrates were synthesized in excellent yields and stereoselectivities to provide only
cis-diol (
4b-
f). On treatment of
4b-
e with BF3-OEt2, pinacol products
5b-
e were obtained in excellent yields and ees.

The only exception was 4f where the alkynyl group could not migrate fast enough, and this led to racemization intermediate B (Scheme 5, vide supra), which ultimately led to loss in ee in 5f. This problem could be gotten around by complexing the alkynyl group of 4f with Co2(CO)6 before subjecting to Lewis acid conditions. Using this solution, after decomplexation, 5f was afforded in high yield over three steps, and in high ee.
After the method was well-established, it was tested in natural product synthetic studies. Isoprenoid-containing natural product, such as
1 was looked at.

Prenyl group installation was performed on (R)-3 using prenylbarium reagent, followed by treatment with BF3-OEt2. The reaction sequence proceeded smoothly, and stereo- and regioselectively to provide 8 in both excellent yield and ee (Scheme 6).