Link: JACS ASAP
Morris Markert, Michael Mulzer, Bernd Schetter, and Rainer Mahrwald*
Institut für Chemie der Humboldt-Universität zu Berlin, Brook-Taylor Strasse 2, 12 489 Berlin, Germany
Morris Markert, Michael Mulzer, Bernd Schetter, and Rainer Mahrwald*
Institut für Chemie der Humboldt-Universität zu Berlin, Brook-Taylor Strasse 2, 12 489 Berlin, Germany
A new method emerged for tertiary amine-catalyzed cross aldol reaction between aldehydes and hydroxyl acetone. This method is different from previous method where tertiary amine was used in conjunction with LiClO4. The initial results are as seen in Scheme 1 of the reactions catalyzed by DBU. In the subsequent scheme, further utilities of the reaction is illustrated, using Hunig's base as catalyst. In some of these reactions, cyclic acetal was obtained along with the expected aldol adduct.
In the subsequent scheme, further utilities of the reaction is illustrated, using Hunig's base as catalyst. In some of these reactions, cyclic acetal was obtained along with the expected aldol adduct. As is readily seen, the reaction predominantly afforded syn-aldol product. The selectivity was probably stemed from the effect of hydrogen-bonding - a welcome complement to the previously known anti-selectivity.
As is readily seen, the reaction predominantly afforded syn-aldol product. The selectivity was probably stemed from the effect of hydrogen-bonding - a welcome complement to the previously known anti-selectivity.
The method is applicable to enolizable aldehyde. The reaction is also regioselective with regards to enolate counterpart, namely only alpha-carbon bearing OH group was observed to add to the aldehyde. In the scheme below, the method was used effectively in the synthesis of furanose 8 and sorbose 9. In addition to DBU and Hunig's base, alkaloid such as cinchonine was also found to be an effective catalyst as demonstrated in the example below.
In addition to DBU and Hunig's base, alkaloid such as cinchonine was also found to be an effective catalyst as demonstrated in the example below.
In addition to DBU and Hunig's base, alkaloid such as cinchonine was also found to be an effective catalyst as demonstrated in the example below.











 The mechanism was proposed to involve addition of NHC to aldehyde to form the extended Breslow intermediate I, followed by reaction of this intemediate with azomethine imine to give intermediate II. This intermediate then tautomerized to give III which intramolecularly acylated to give product 3.
The mechanism was proposed to involve addition of NHC to aldehyde to form the extended Breslow intermediate I, followed by reaction of this intemediate with azomethine imine to give intermediate II. This intermediate then tautomerized to give III which intramolecularly acylated to give product 3.

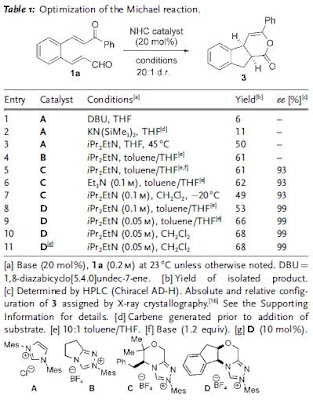
 The reaction tolerated various R groups on aldehyde including aliphatic (entry 6) and extended olefin (entry 7). However, when R group was eletron-withdrawing group-substituted phenyl ring, the reaction did not yield any product (entry 8).
The reaction tolerated various R groups on aldehyde including aliphatic (entry 6) and extended olefin (entry 7). However, when R group was eletron-withdrawing group-substituted phenyl ring, the reaction did not yield any product (entry 8). Further transformations of pyridazinone 4 were found to be facile both with MeOH and BnNH2 to give the corresponding ester 19 and amide 20 in almost quantitative yields.
Further transformations of pyridazinone 4 were found to be facile both with MeOH and BnNH2 to give the corresponding ester 19 and amide 20 in almost quantitative yields.