Link: JACS ASAP
Haifeng Du, Weicheng Yuan, Baoguo Zhao, and Yian Shi*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523
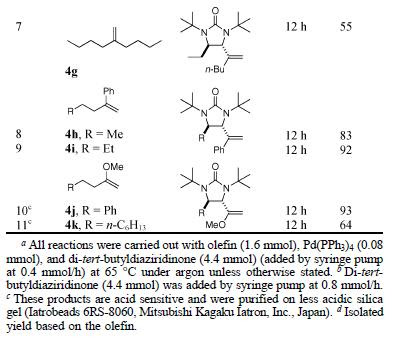
This is a new method di-amidation of terminal alkenes at the allylic and homoallylic positions using di-tert-butyldiaziridinone (2). A similar method was reported before with conjugated diene 1. In this method, terminal alkene 4 was used to give similar product.
The reaction is applicable to a variety of alkenes, affording products with trans-stereochemistry in modest to excellent yields.

The products of the current reaction is useful in further transforming to give 1,2-diamines such as 6.

In addition to mono alkenes, the reaction was also applied to bis-terminal alkenes. In case of 7, the products were formed as a mixture of 8a and 8b in a 1:1 ratio. In case of 9, while 11 was formed, both 10a and 10b were isolated and are believed to be intermediates in the reaction. Both 10a and 10b when subjected to Pd(PPh3)4 transformed give 11.


The mechanism of the reaction was proposed to be as followed:

Essentially, the reaction probably goes through intermediate diene 15 formed in situ. A more detailed mechanistic study is needed.





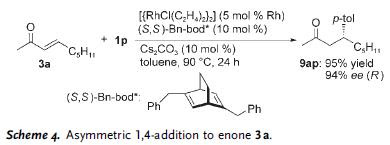

















 The reaction was also shown to be stereospecific, that is the stereochemistry in the starting material was effectively transferred to the product as shown in the scheme where stereochemistry of carbon bearing the phenyl group in 1a was retained in product 5a.
The reaction was also shown to be stereospecific, that is the stereochemistry in the starting material was effectively transferred to the product as shown in the scheme where stereochemistry of carbon bearing the phenyl group in 1a was retained in product 5a.















 It should be noted in entry 5 that when ortho-substituted aromatic group of imine 21 was used, the reaction occurred diastereoselectively to give a 4:1 atropisomeric mixture.
It should be noted in entry 5 that when ortho-substituted aromatic group of imine 21 was used, the reaction occurred diastereoselectively to give a 4:1 atropisomeric mixture.



